The Boltzmann Maps web app now employs GPU-accelerated OpenMM software for compound energy minimization in the context of a protein. The reported energies include van der Waals and electrostatic energies between compound and protein, as well as the change of a compound’s internal energies between the unbound and bound configurations (stress). These energy reports are a key metric for evaluating and comparing compounds and modifications. OpenMM integration allows Boltzmann Maps to provide this data with improved quality and speed.
OpenMM is an open-source toolkit for molecular simulation. It is highly flexible with its custom functions and has high performance, especially on recent GPUs. More information can be found at: https://openmm.org.
Energy minimization in BMaps
In BMaps, when a compound is imported, docked, or modified, it is generally desirable to energy minimize the compound. This typically takes only a few seconds, depending on the size and complexity of the compound. Forcefield parameters and partial charges are automatically determined for the compound. The energy minimization is invoked with a single click of the Energy Minimize button found in the Modeling Tools button tray.
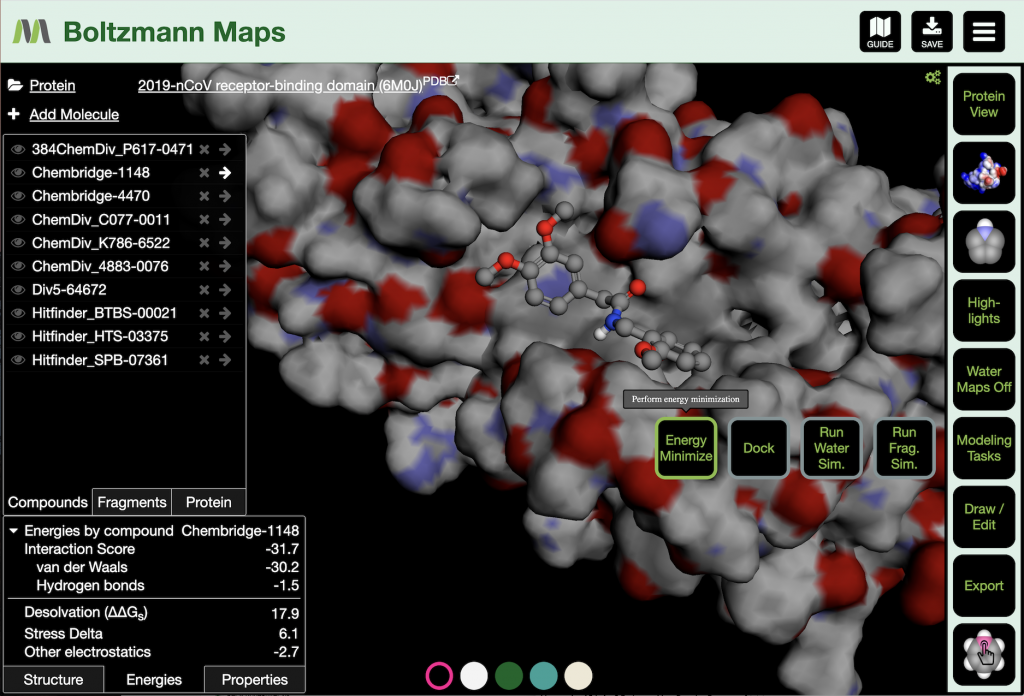
After minimization, the energy report is found in the lower left corner of the BMaps window. The Interaction Score incorporates the contributions of van der Waals and hydrogen bond energies. The total energy of the electrostatic interaction between the compound and the protein is factored into a Hydrogen bonds term and Other electrostatics (longer range, distributed interactions). The Stress Delta term is the change of the internal energy of the compound between its unbound and bound conformations. Stress includes both bonded energy terms (bond stretching, bond angle bending, and torsion twisting) and internal van der Waals and Coulomb interactions. Finally, the Desolvation (ΔΔGs) term shows the energetic cost of removing water from the compound and the protein binding site to allow binding.
When optimizing the binding of a compound, it is useful to keep an eye on these energy terms as changes are made to the compound. Compound modifications include fragment growing and terminal replacement from the 3D workspace (right-click on a compound atom), as well as edits from the 2D sketcher (Edit Active Cmpd button in the Draw/Edit button tray). Generally, compound-protein binding is an interplay between the attractive van der Waals energy and the desolvation energy penalty, as these are often the largest contributors to binding. Hydrogen bonds enhance the binding and provide specificity.
BMaps’ OpenMM Implementation
Energies directly calculated with OpenMM comprise the van der Waals, stress, and total electrostatic energy components. The specific contribution of hydrogen bond energy and the cost of desolvation are calculated by BMaps.
Boltzmann Maps’ implementation of OpenMM currently runs on a custom GPU server. The BMaps team is also investigating the feasibility of migrating the web app’s OpenMM execution to the browser with WebAssembly and WebGPU. Stay tuned!
The current implementation uses a rigid protein during minimization, but OpenMM does support flexible relaxation of the protein as well as the compound. We anticipate supporting this in the BMaps interface in the future.