Intelligent ADMET for Intelligent Design
Drug discovery has entered a new era, defined not only by how rapidly we can generate new molecules, but by how intelligently we can evaluate them. With our latest update to Boltzmann Maps (BMaps), we’re bringing that intelligence one step further by incorporating MapLight Therapeutics’ ADMET property prediction tools directly into BMaps.
This integration enables researchers to analyze molecular designs made in BMaps with a holistic view of their pharmacokinetic potential. The improved potentials makes it easier than ever to identify compounds that are not only potent, but also likely to behave as real drugs in biological systems.
MapLight ADMET
MapLight represents one of the more accurate and rapid AI-driven frameworks for predicting ADMET (Absorption, Distribution, Metabolism, Excretion and Toxicity) properties across diverse chemical space. You can read more about their performance on benchmarks in Reference (1) cited below.
The platform employs a combination of random forest models trained on extended-connectivity fingerprints (ECFP)and gradient-boosted decision trees (GBDT) that leverage over 200 molecular descriptors — from topological indices and polar surface area to atom-level physicochemical features. This hybrid approach enables MapLight to capture both structural similarity patterns and nonlinear feature relationships, leading to predictive accuracy across a wide variety of ADMET endpoints.
Within the Therapeutic Data Commons (TDC) Leadership Boards, MapLight’s ADMET predictors rank among the top three performing models for key endpoints such as P-glycoprotein (P-gp) inhibition and blood–brain barrier (BBB) permeability. These properties are essential for understanding whether compounds can reach their target sites effectively or pose drug-drug interaction risks.
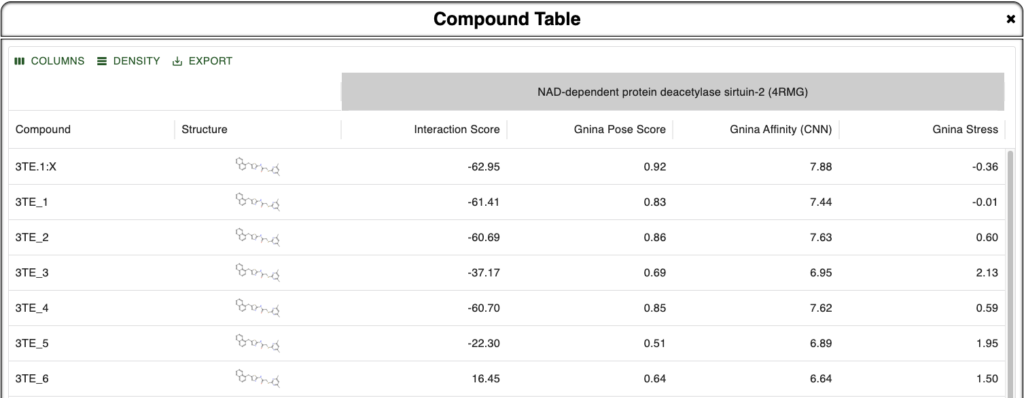
Incorporating MapLight into BMaps provides users with seamless ADMET prediction capabilities alongside existing design and scoring workflows. Over 20 different ADMET properties are automatically predicted as compounds are imported alongside their crystal protein, or else created in the BMaps platform. No matter what stage in the drug design process you are in, ADMET scoring can now be run in a single step. Examples of various views of ADMET properties shown in the figures below:



Why Does This Matter?
In silico design has long focused on potency — optimizing for how tightly a molecule binds to a target protein. But as every medicinal chemist knows, potent molecules aren’t always good drugs. Issues like poor solubility, rapid metabolism, or efflux by transporters can derail otherwise promising leads.
By integrating ADMET prediction directly into BMaps, we enable users to prioritize compounds that balance potency with druggability.
This means:
- Fewer late-stage failures due to pharmacokinetic liabilities
- More efficient lead optimization cycles
- Smarter allocation of resources
The ability to predict liabilities before synthesis or experimental testing represents a major step toward democratizing drug design. This improvement of resources helps both academic and industry researchers make informed decisions earlier in the pipeline.
The addition of MapLight ADMET scoring is only a part of a broader effort for BMaps: to make drug discovery accessible, interpretable, and actionable for every researcher.
Check it out in BMaps today at Bmaps.app
Citations:
(1) Notwell, J. H.; Wood, M. W. Admet property prediction through combinations of molecular fingerprints. arXiv preprint arXiv:2310.00174 2023.
(2) Huang, K.; Fu, T.; Gao, W.; Zhao, Y.; Roohani, Y.; Leskovec, J.; Coley, C. W.; Xiao, C.; Sun, J.; Zitnik, M. Artificial intelligence foundation for therapeutic science. Nature Chemical Biology 2022, 18 (10), 1033-1036. DOI: 10.1038/s41589-022-01131-2.