Quick Start Guide
This Quick Start guide will walk you through the basic steps of using Boltzmann Maps:
- Adding your target structure and compound
- Modifying the compound and observing the effects on the chemical properties.
- Exporting your results
- (Advanced) Running new water and fragment simulations
To get started, click the login button to create an account and subscribe to the service. You can change the service level at any time.
When Boltzmann Maps loads, the Choose protein list will appear, containing hundreds of prepared structures with precomputed water and fragment data.
If we don't have your target, you can import it from the PDB or load your own pdb file.
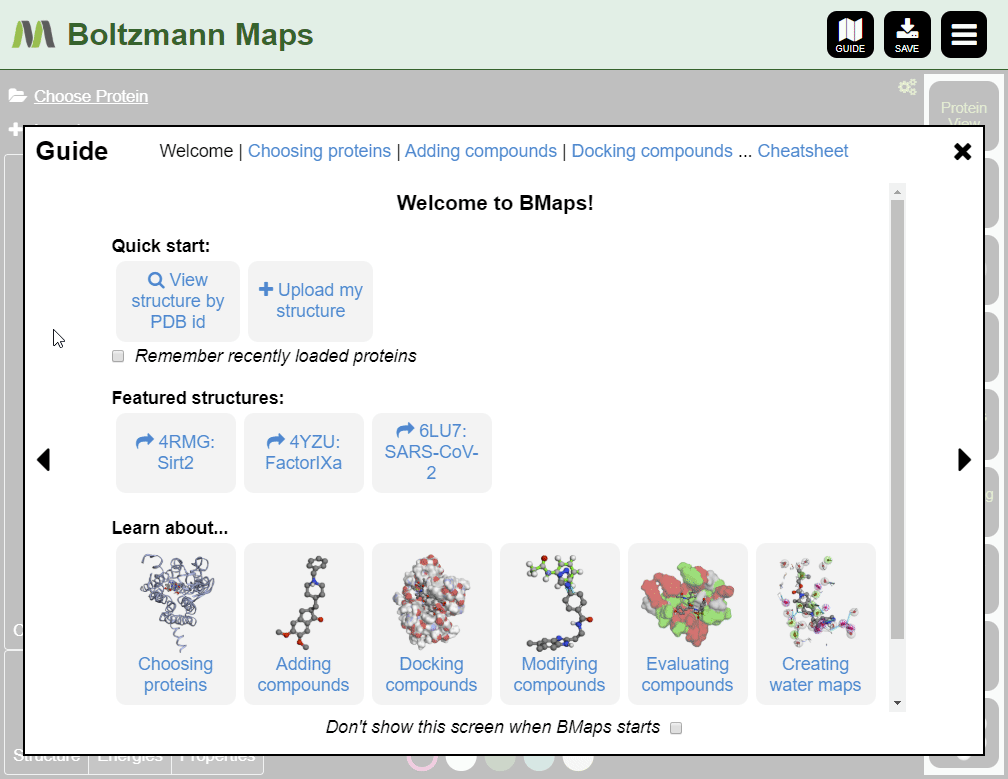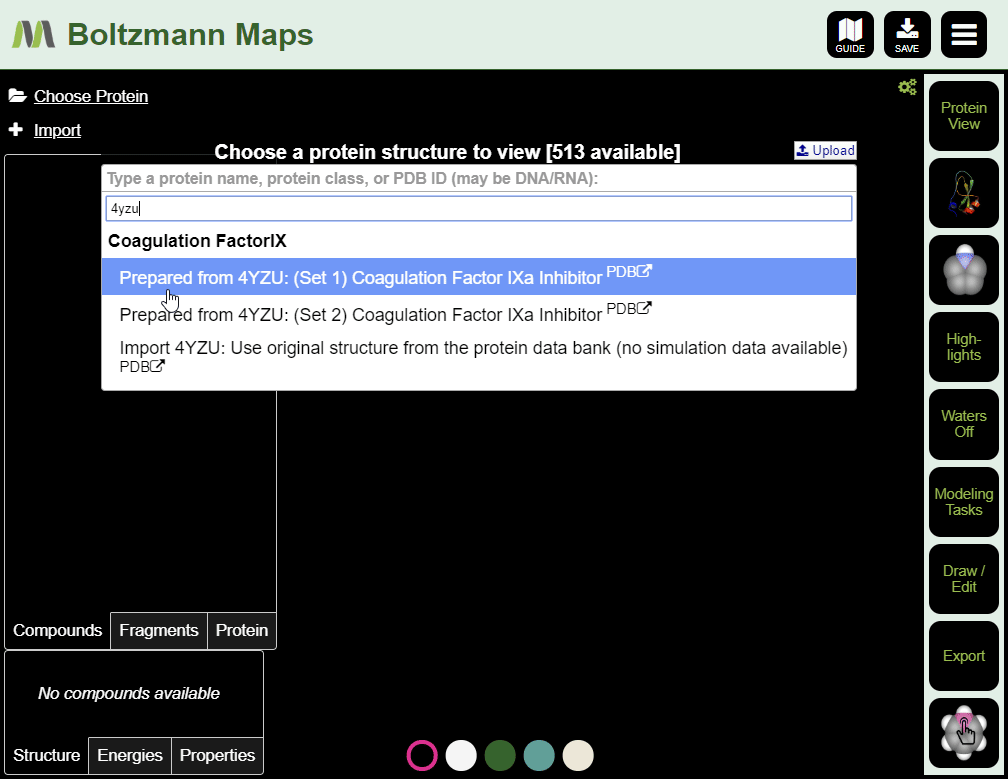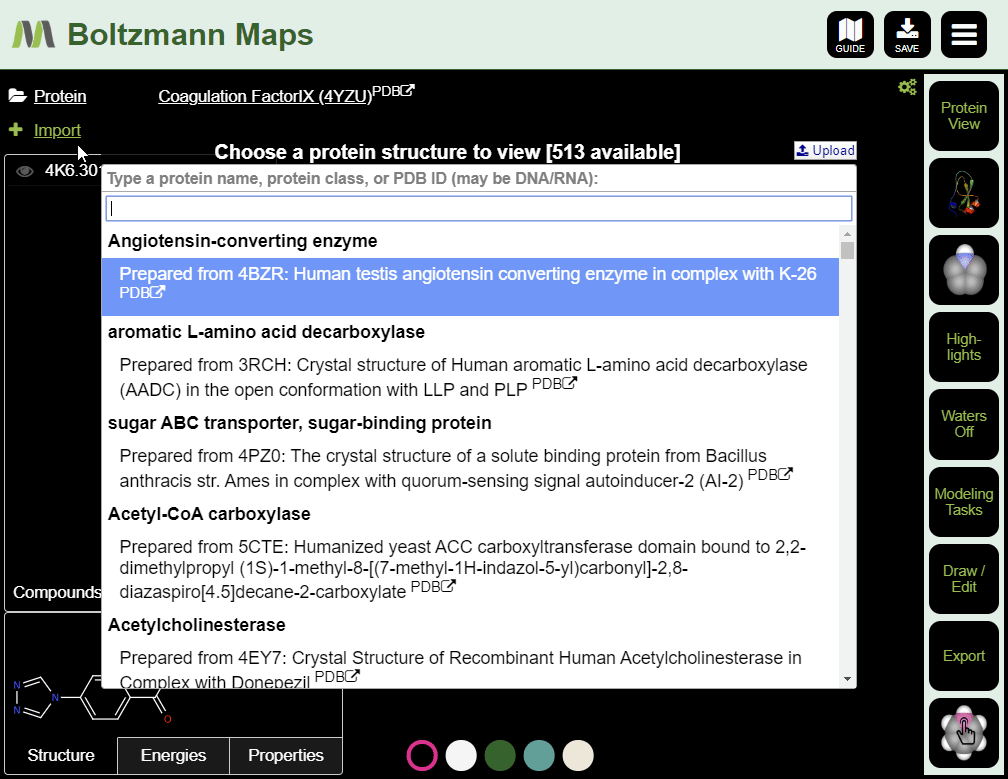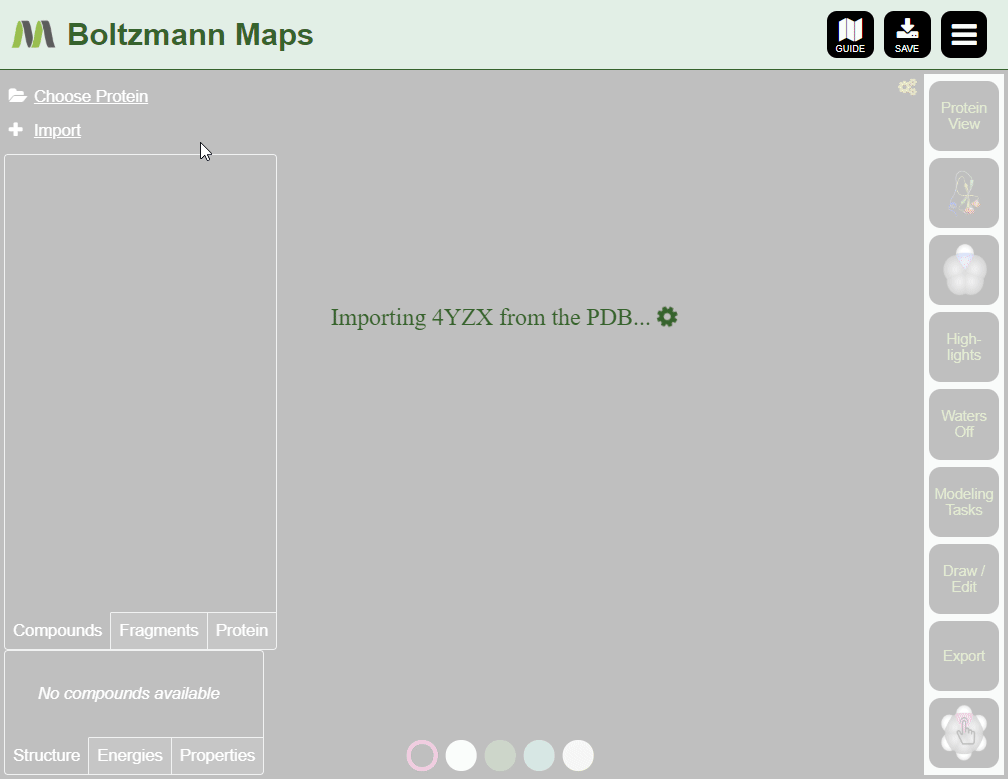
To load by PDB id:
- Enter the pdb id of your target protein into the protein list.
- Click the full protein name to load a prepared structure, including water and fragment data.
- Click "Import pdb id from the protein data bank" to load the structure only; water and fragment data will not be available.
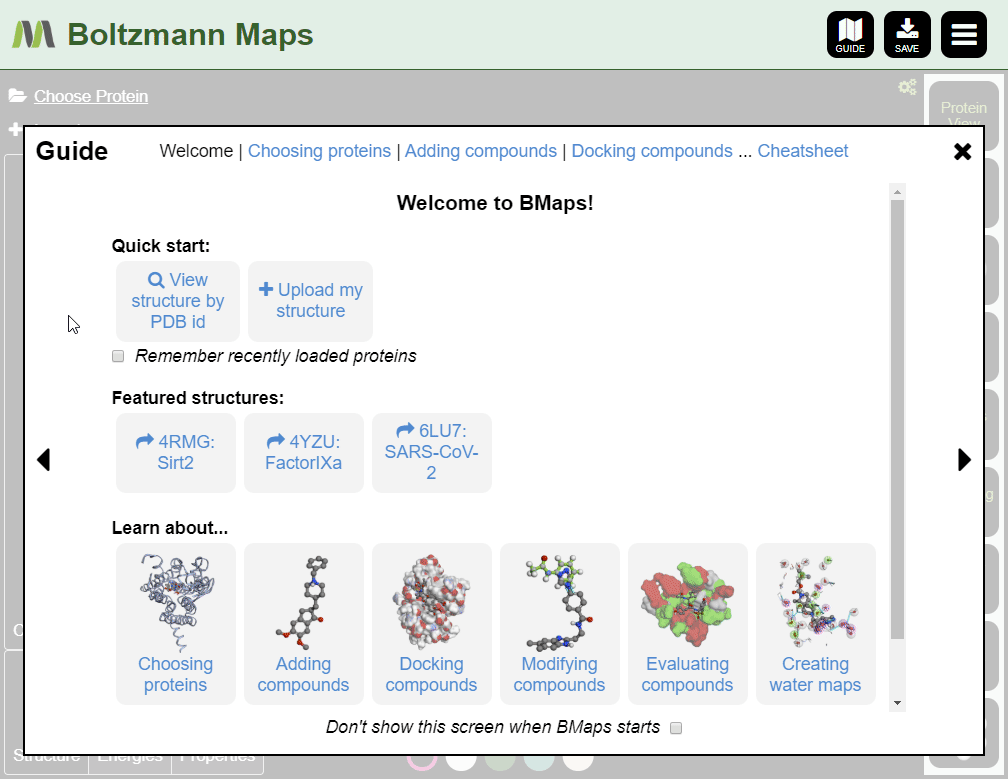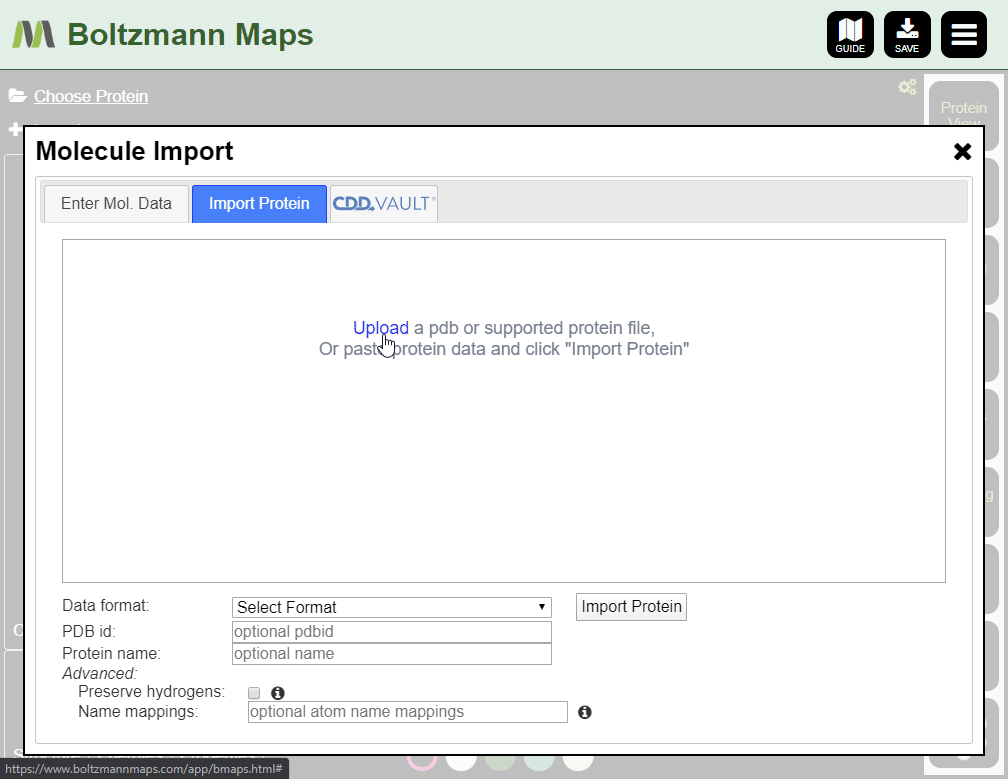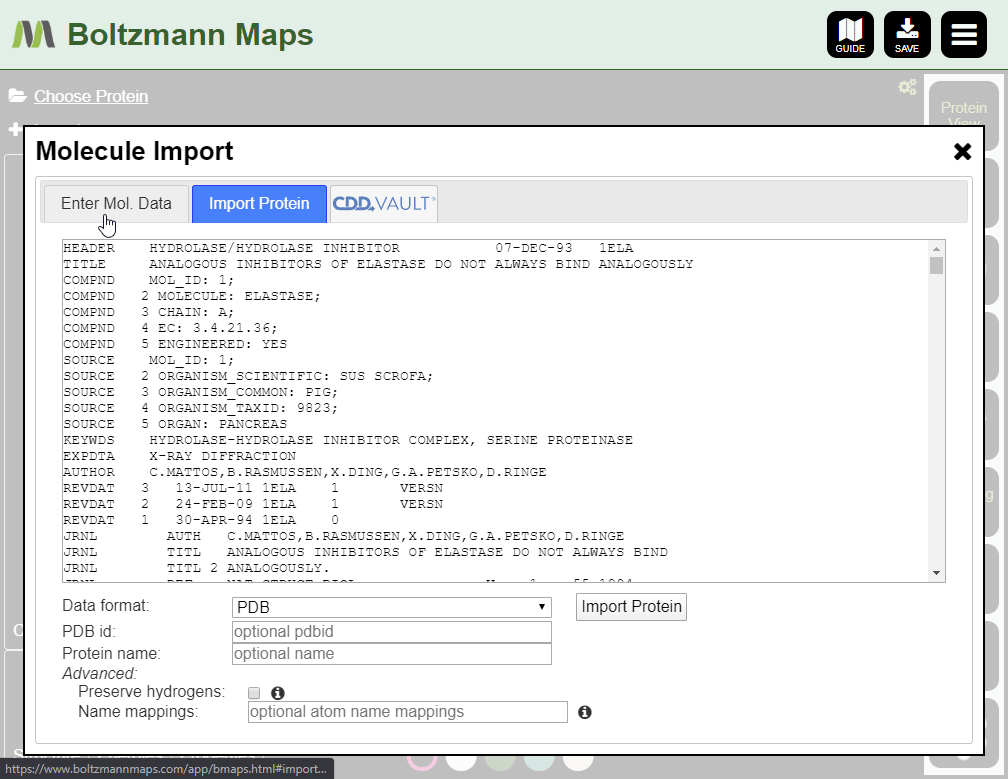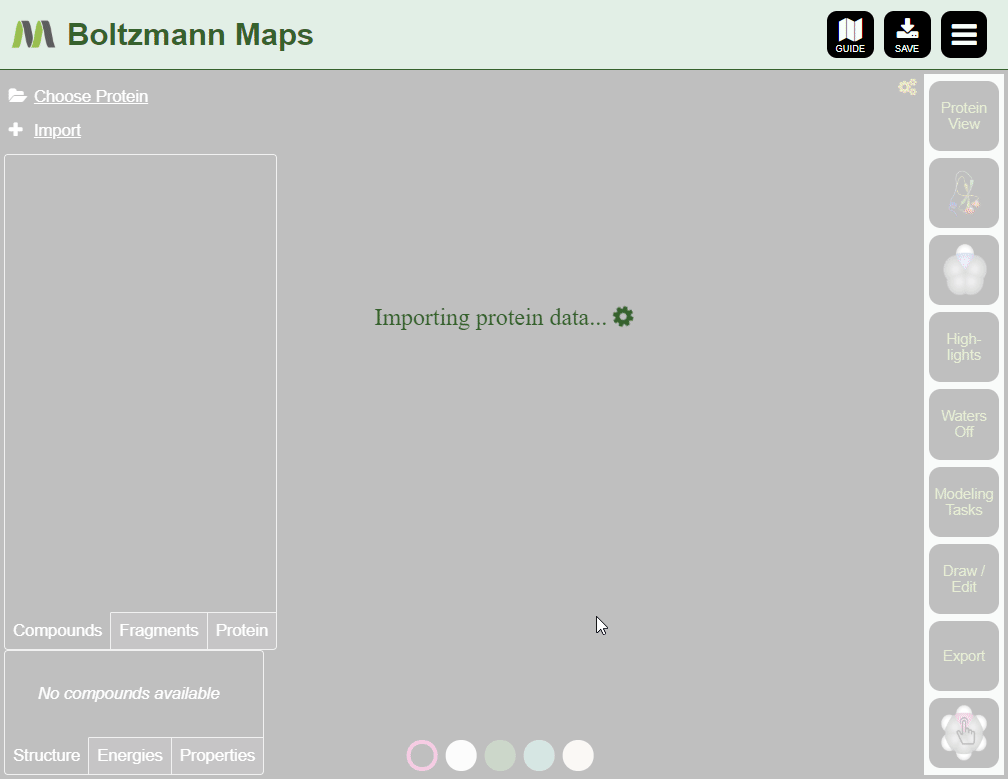
To load your own protein, open the Import tool and go to the Import Protein tab to enter data or upload pdb files.
There are several ways to add compounds:
Add a file or compound data
- Drag and drop a file into the workspace
- Paste compound data into the workspace
- Open the Import tool and use the Enter Mol Data tab to enter compound data or upload files
- Supported formats: cdx, sdf, smi, mol (v2000), mol2, pdb, cif, cdxml, cml
Use the 2D sketcher
Choose Draw New Cmpd from the Edit menu and draw a new compound.
Import from CDD Vault
- Open Import tool and use the CDD Vault tab to add your API key and connect to your Vault.
- Select your vault and project to view your stored compounds.
- Mouseover your desired compound and click or if you want to preserve coordinates.
Docking new compounds
When you load a compound, the default behavior is to align the compound to a crystal ligand. If there is no crystal ligand, or if you want a better pose, dock the compound before doing any design.
The Dock control is in the Modeling Tasks menu.
To view important chemical properties like interaction energies and logP, click the Energies and Properties tabs in the bottom left.
To visualize energies semi-quantitatively, especially the cost of desolvation, see the Highlights menu.
To view crystal waters or highlighted water maps, open the Waters. View hotspot clusters from the Highlights menu.
To view fragment map summaries, visit the Fragments tab of the left side selector. Click a fragment to view the summary. A slider will appear, allowing you to filter out fragments that don't meet the energy threshold.
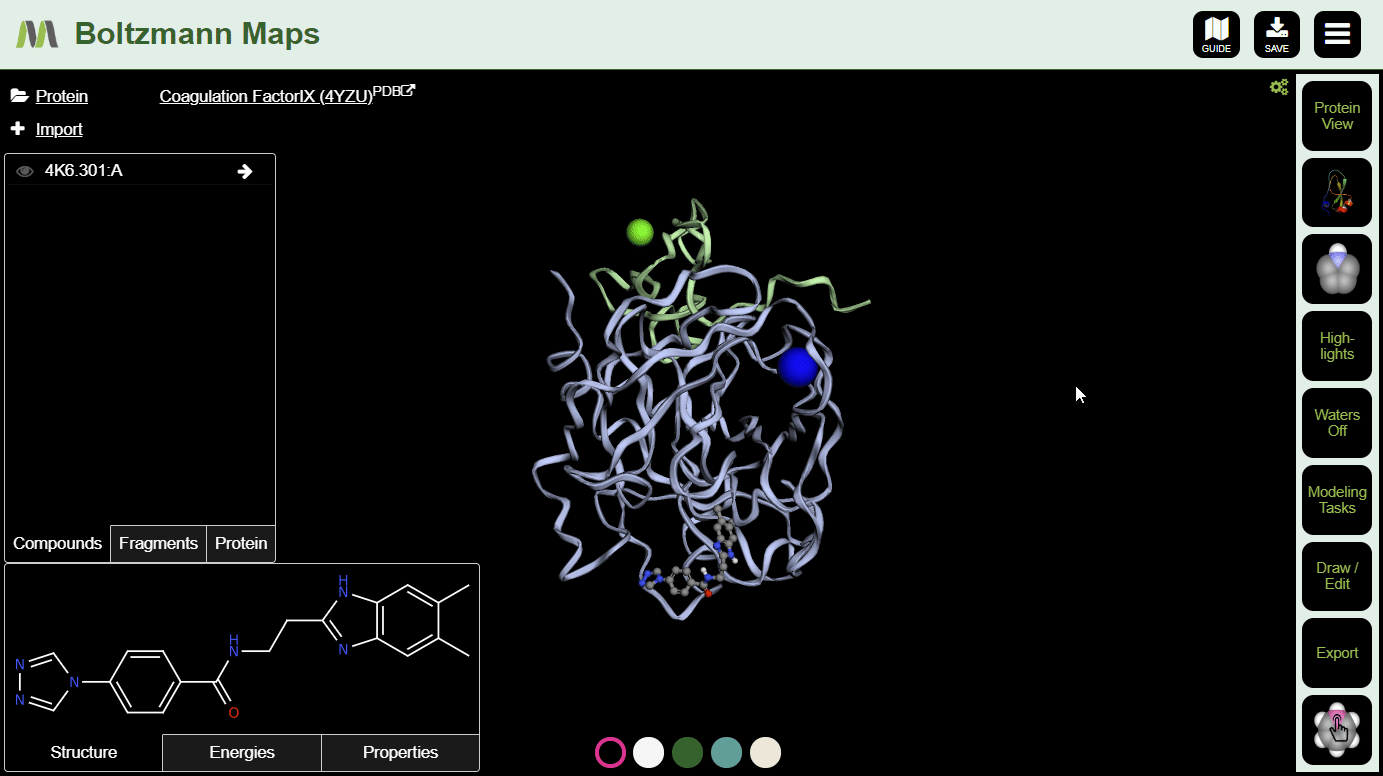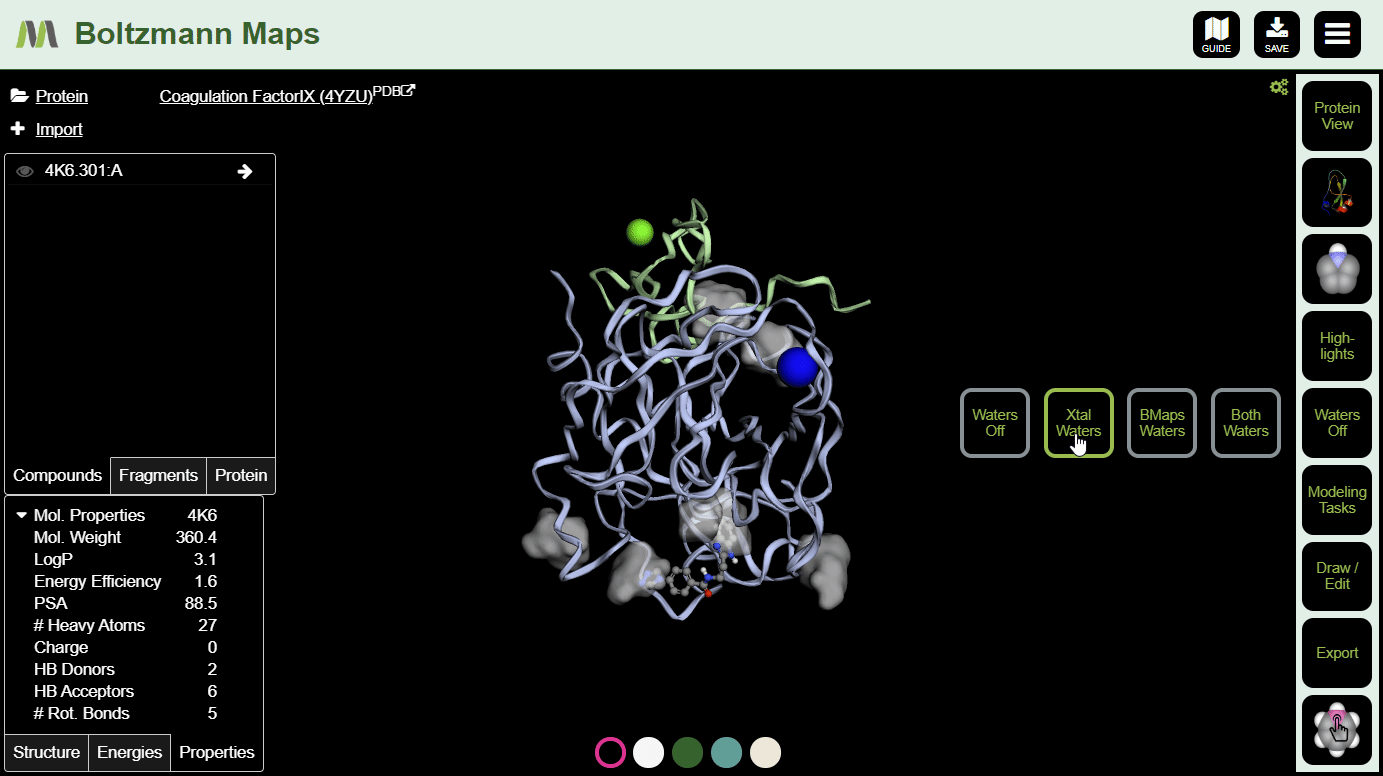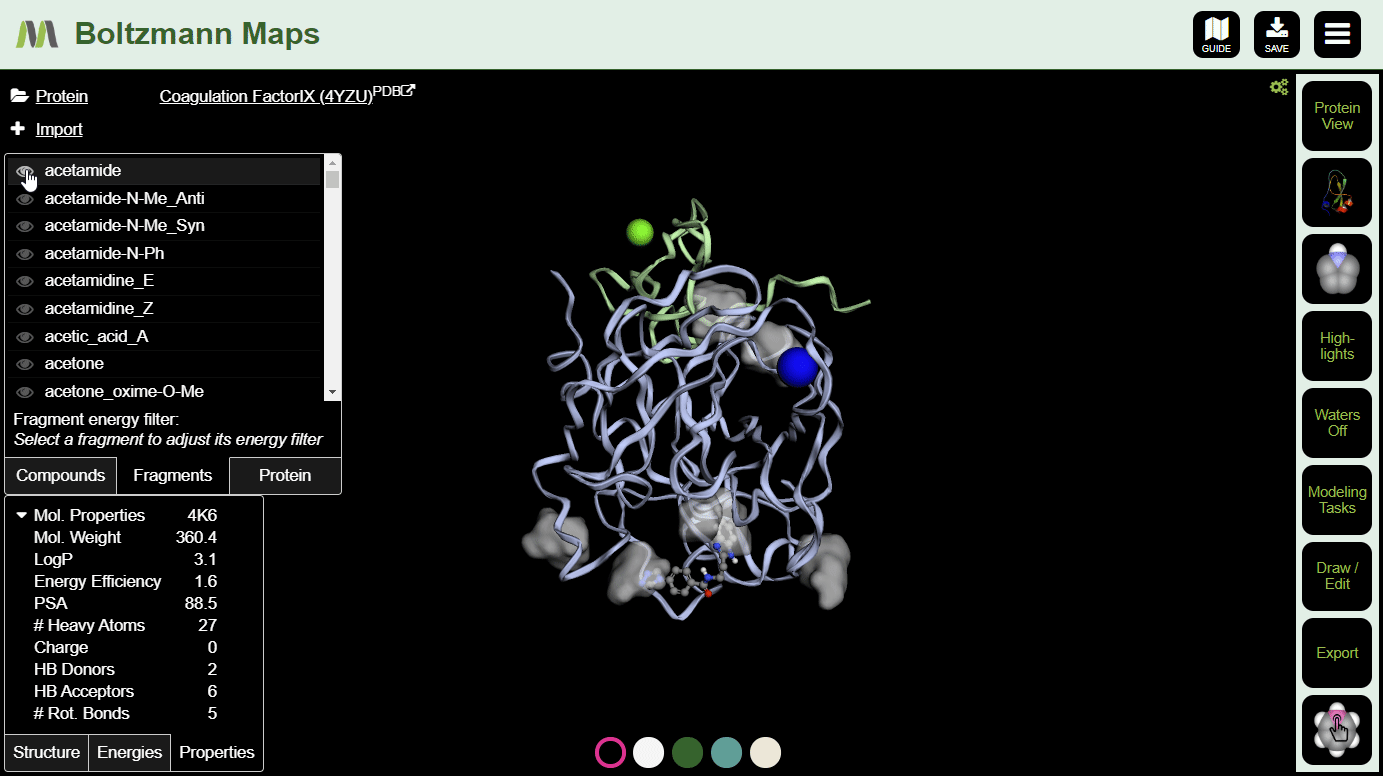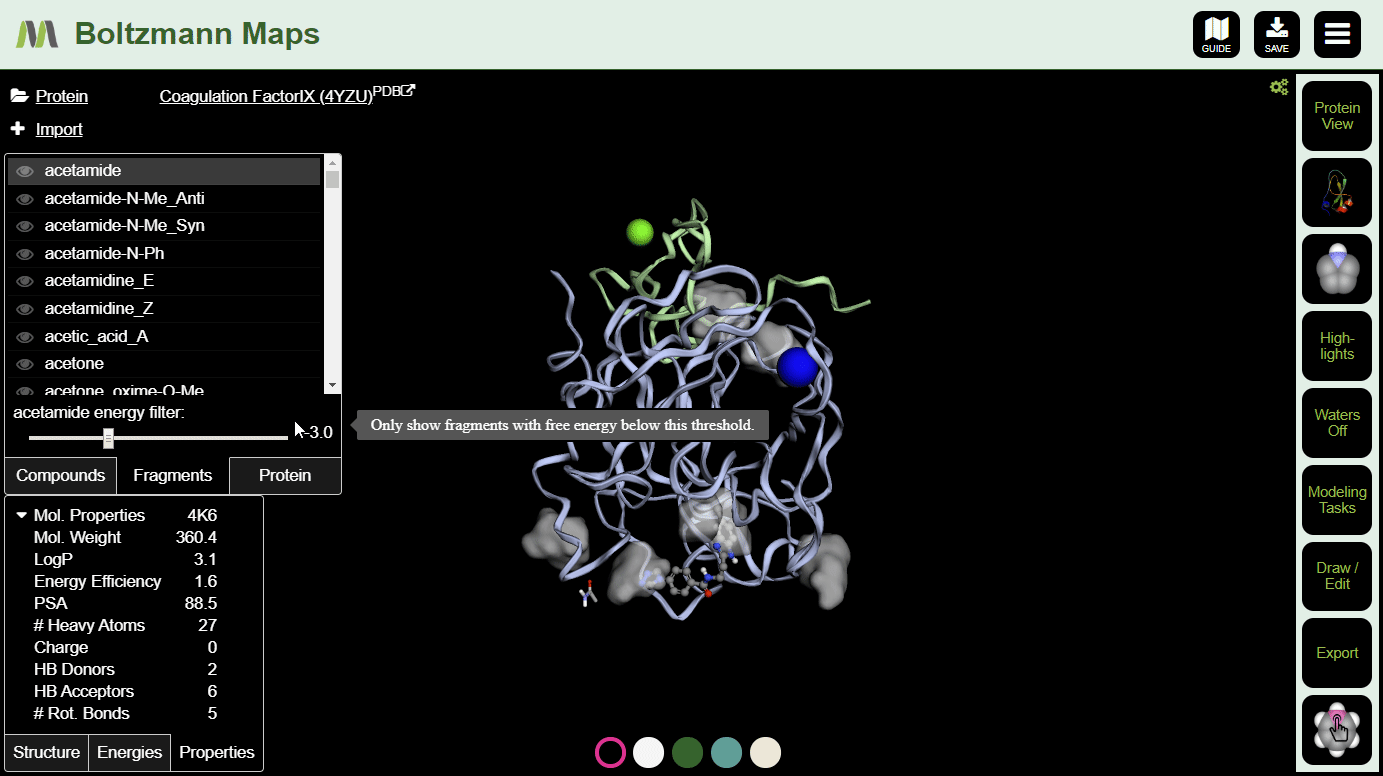
Now that your compound is in position and you have baseline energies, it's time to make some modifications and see if they improve the compound's chances.
There are several ways to modify compounds:
- 2D Sketcher:
Choose Edit Active Cmpd. from the menu. - Terminal replacement:
Right-click an atom in a terminal group and choose a replacement from the Replace terminal section. - Growing to fragments:
Right-click an atom and click Bond link from the Grow section. If the structure came with fragment data, then nearby fragments will appear as candidates to replace a section of your compound.
After making a few changes, click the s on a few compounds in the left-hand compound list. Then you can compare the energies and properties in the tables below.
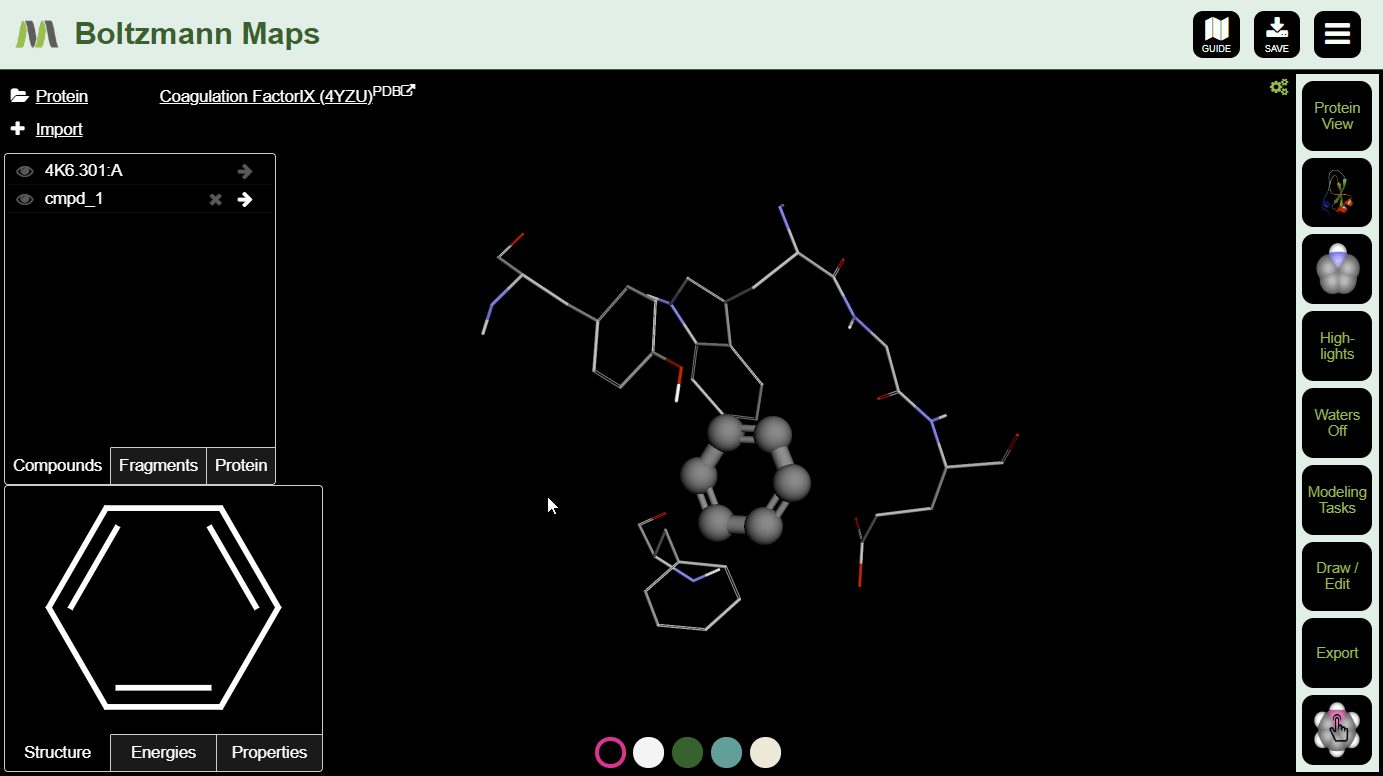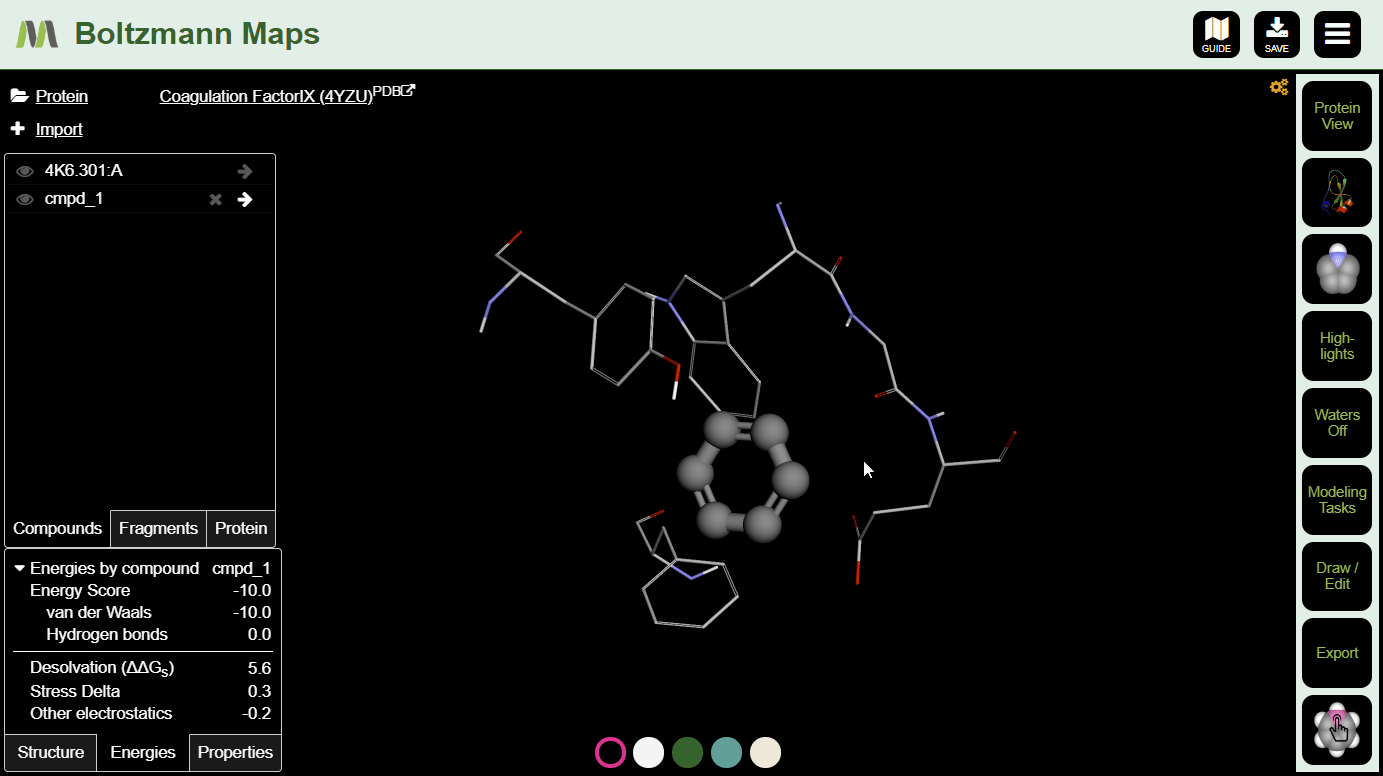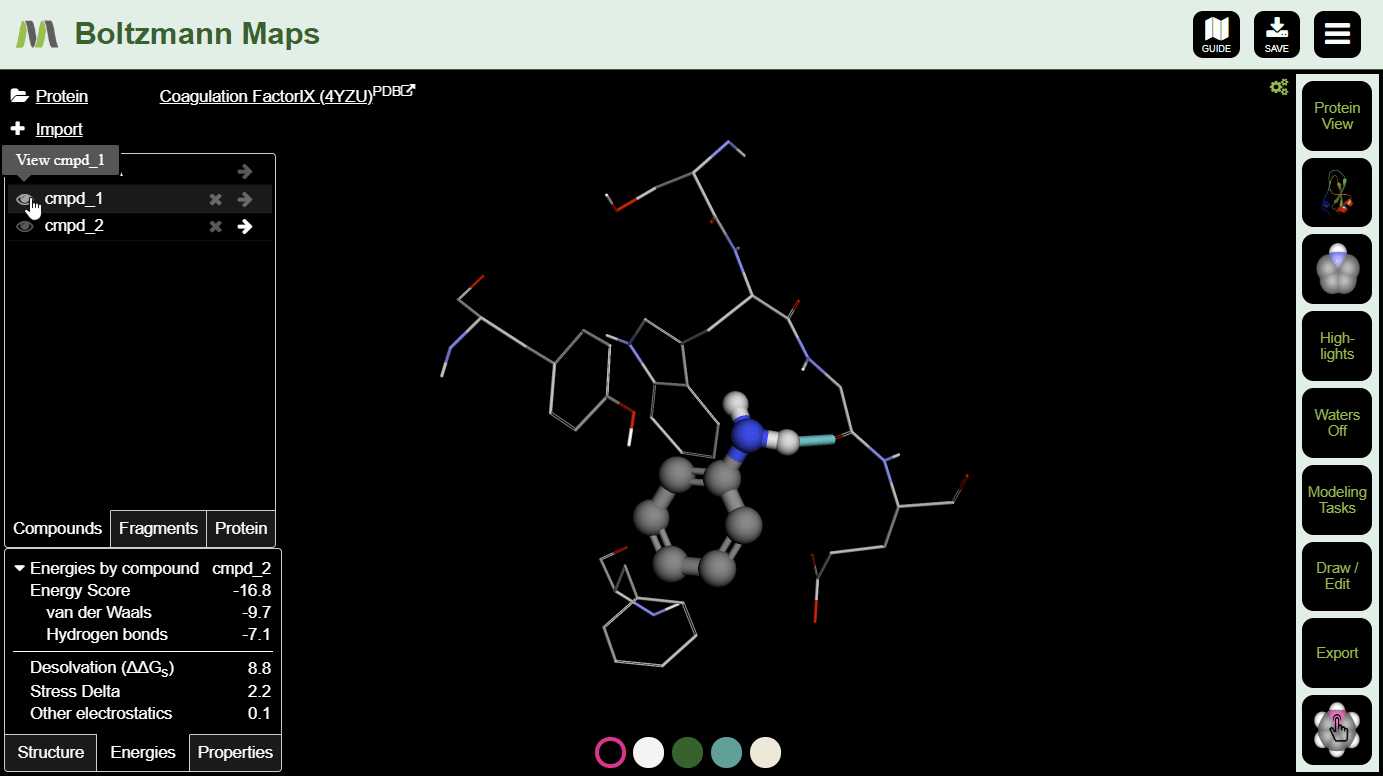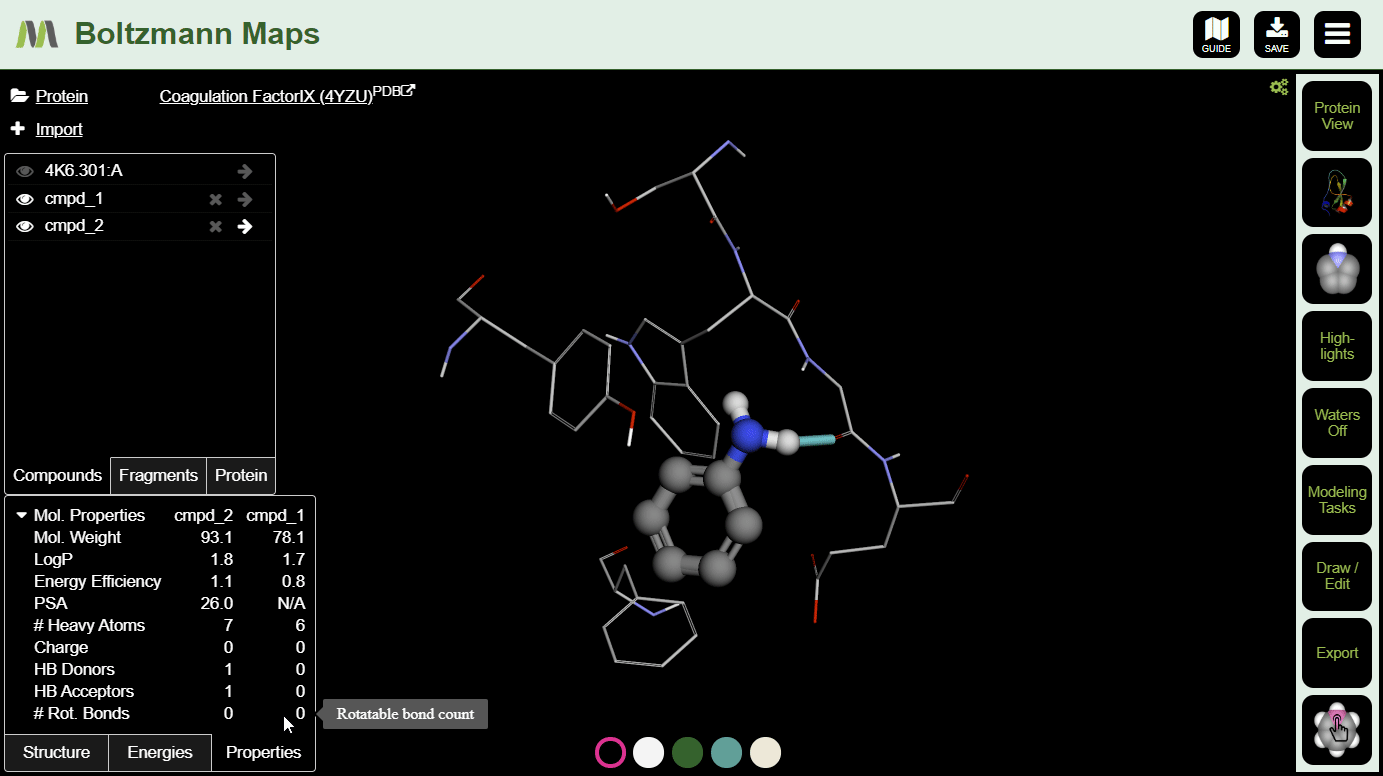
When you are ready to move your compound designs along the pipeline, use the tool to:
- Download your data in a number of common formats
- Search PubChem for similar compounds
- Upload your compound to CDD Vault, if you have an account.
In order to save everything and pick up where you left off later, click the button in the top right to download a session file with your protein and all your compounds. Your compounds aren't stored in our server.