Boltzmann Fragment Maps
Fragment maps derived from Grand Canonical Monte Carlo (GC/MC) simulations are an important source of information for drug discovery and life sciences.
Maps of where small chemical fragments bind to proteins are proving to be an important source of information for life sciences research, most particularly in drug discovery, as has been well documented.1-4 For example, such maps are used to:
- Discover binding “hot spots” by fragment clustering;
- Identify how fragment binding patterns differ among protein structure variations, critical for achieving selectivity, mutation avoidance, or multi-targeting;
- Assess the impact of tightly-bound waters on biological protein function and ligand binding;
- Find starting points for difficult targets;
- Generate ideas for modifications in lead optimization.
Methods for deriving fragment maps can be compared based on the information they provide and the relative cost in obtaining them. The need is to develop the best (most comprehensive and predictive) information about fragment binding for the lowest cost. Further, these maps need to be quickly and easily accessible. Using cheaper, less accurate information results in more false positives, a larger number of compounds to make and test, and fewer opportunities to evaluate compounds with a range of properties. Until now the best fragment maps have not been generally available, being captive to proprietary enterprises or limited by the operational complexity and expertise required in producing them.
What are the best fragment maps?
Experimental approaches to obtaining fragment maps by X-ray crystallography or NMR, while useful, are often prohibitively expensive for the average drug discovery company or academic research groups. Also there are practical limitations to the number and type of fragments can be tested this way (e.g. solubility, reactivity, lipophilicity, minimum size). The information is limited to the binding pose, restricting potential applications. On the positive side, crystal structures provide real information about the protein conformation seen by bound fragments.
Computationally-derived fragment maps are more general, low cost, and admit smaller fragments (allowing for fine-grain design), but have traditionally not been very predictive. The key reasons for a lack of predictability of fragment binding affinity are the difficulty of accounting for configurational entropy and solvation contributions (ΔΔGs) to the binding free energy. In some cases, protein flexibility is a challenge. Such maps can be segmented into those developed by probing or docking, and those produced by molecular simulations (see review by Loving et5 al, Schrodinger, Inc.).
Over the last decade, we have been evolving an alternative approach: low-cost fragment maps derived from Grand Canonical Monte Carlo fragment-protein simulations incorporating simulated annealing of chemical potential (GC/MC-SACP), conceived by Guarnieri.5-7 Fragment maps generated this way (Boltzmann Maps or BMaps), with rigorous statistical physics, allow ranking of binding sites by relative excess chemical potential (average free energy per fragment). This free energy metric includes desolvation free energy8 and configurational entropy.7 This is in contrast to ad hoc scoring functions used by others. Unlike other methods, the GC/MC-SACP method provides a statistically accurate Boltzmann distribution of fragment poses. This is important information for assembling fragments into ligands or understanding water binding.
The advantages of annealing chemical potential are:
- More comprehensive sampling;
- The ability to rank binding sites by the lowest chemical potential at which they are at least 50% occupied.
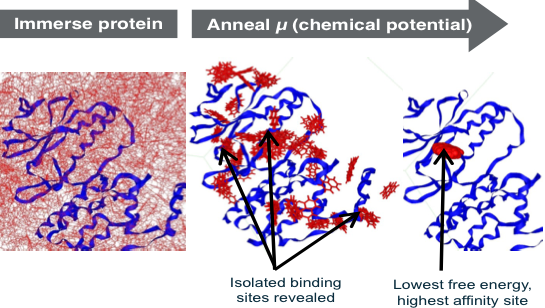
The better sampling occurs because, at high positive chemical potentials, fragments can temporarily occupy energetically unfavorable positions while they move to explore optimal sites, rapidly overcoming energy barriers to reach low energy states (Figure, left panel). At low negative chemical potentials, most of the surface of the protein is evacuated and binding pockets become isolated, and are thus separately characterized by the excess chemical potential, and thus average free energy for ranking (Figure, right panel). Using GC/MC-SACP, the water maps are unique in efficiently discovering multi-body water interactions involving two or more waters—a limitation of other commercial and free water maps. In the 2011 SAMPL3 competition (OpenEye), the performance of our GC/MC-SACP in ranking fragments was documented.15
While the quality of GC/MC-SACP derived maps are generally acknowledged to superior to other maps, they have historically been considered to be expensive to produce. Compared to earlier GC/MC codes, we have extended the GC/MC-SACP with a variety of new sampling algorithms, efficient electrostatics modeling, and adaptive annealing strategies that have drammatically reduced the cost ($.10-$.50 per map on AWS EC2) while improving the accuracy of the sampling.
In the last several years, we have successfully applied these maps in over a dozen lead identfication programs (including initial hits on PCSK9 and RecA – protein-protein interactions) and several lead optimization campaigns—novel nanomolar compounds, among < 40 synthesized, for ACCase, 11BetaHSD-1, renin, DHFR, and others. Over 250,000 simulations, on >100 proteins of various classes validate the generality of the method. To our knowledge, no other method has been operated at this scale in the production of fragment and water maps. Using these maps in successfully identifying and optimizing drug lead compounds on multiple targets validates their relevance to experimental outcomes.