Today we introduce real-time quantification of π-π interactions in BMaps using quantum mechanically accurate methods. π-π interactions play a crucial part in the stabilizing and selectivity of protein-ligand interactions for drug design. The aromatic amino acids (Phe, Tyr, and Trp) are common interacting species in active sites, and as any examination of recently approved drugs will tell you, aromatic rings are key components of effective drugs for both interactive and structural reasons.
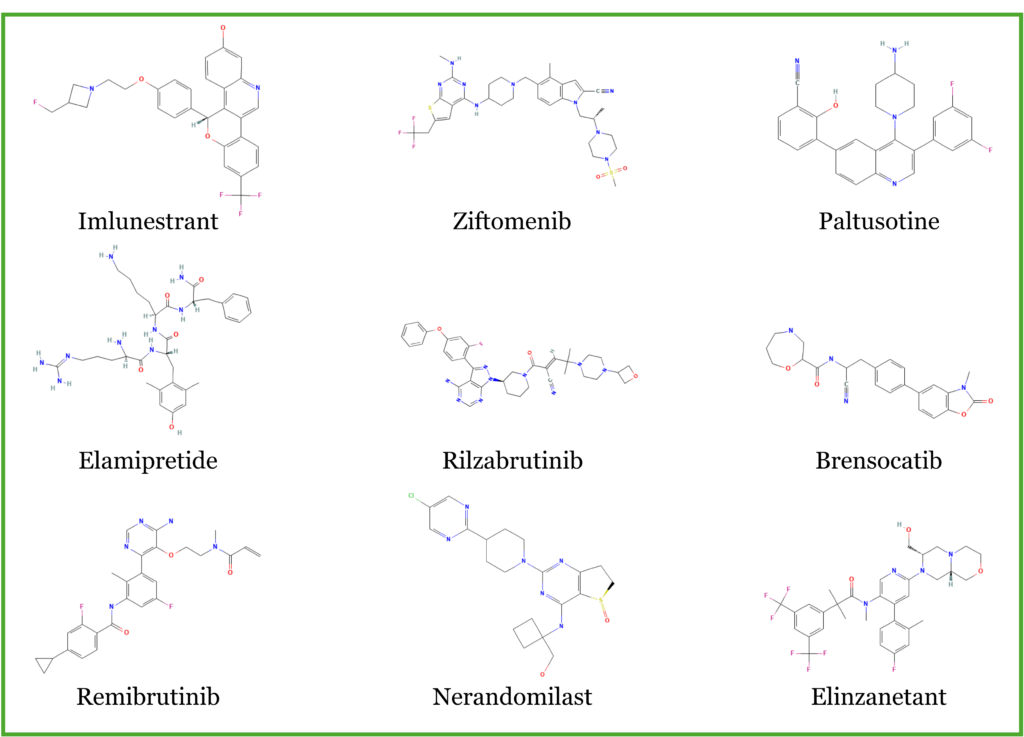
Figure 1. Subset of 2025 FDA approved drugs with aromatic rings
For example, non-nucleoside reverse transcriptase inhibitors are known to interact selectively with HIV-1 reverse transcriptase, but not HIV-2 reverse transcriptase. As studies have shown, the HIV-1 Tyr181/Tyr188 create an aromatic cage while HIV-2 lacks this same environment. Mutation studies in HIV-1 have shown that the loss of this aromatic cage knocks out this selective binding [1, 2, 3]. Furthermore, cases like a study of Imatinib with and without inclusion of quantum mechanical (QM) energy terms in the description of protein-ligand interactions have shown 10’s of kcal/mol inaccuracy versus experimental binding energetics. When the QM energy terms are included, this error goes away. Such results make all but impossible to determine rational design trends. [4] It is for this reason that accurate quantitative understanding of the pi-pi interactions created by these aromatic rings at the quantum mechanical level is so necessary for designing new small molecules drugs.
Current Standard of Practice – Computational Scaling Prevents Quantification for Large Systems Like Proteins With QM…Or At All
Despite this importance, current methodologies of quantifying π-π interactions take days to weeks for a single calculation, often requiring simplification of the model system to perform the calculation. Therefore, until now, quantifying π-π interactions in drug design campaigns has been impractical. Today, Conifer Point’s BMaps is changing that with the inclusion of atomistically separable quantum mechanical free energy prediction of π-π interactions. Our internally developed machine learning models are trained with density functional theory with long-range dispersion corrected functionals and complete electron wavefunction basis sets. With these quality models we can construct atomistic pictures of protein ligand interactions across the entire protein in seconds. This capability is currently not available in any other computational chemistry software or machine learning models in the literature. The key reason for this uniqueness lies in the atomistic separability of our models which will be discussed in an upcoming manuscript.
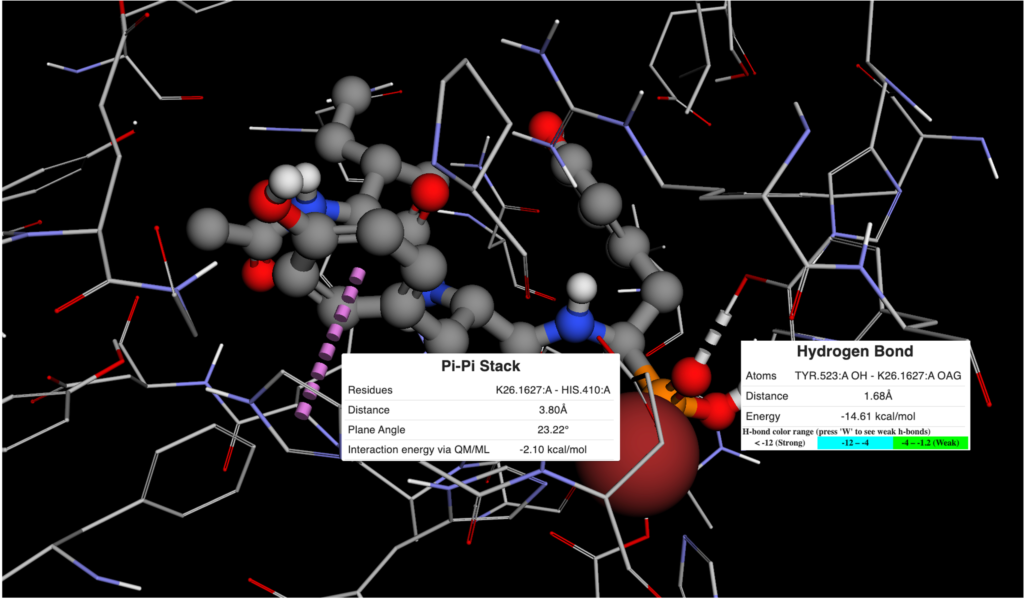
Figure 2. Visualization of π-π and Hydrogen Bonds Between 4BZR and K26 in BMaps.
See the Interactions You’ve Been Missing.
Try the π-π Interaction Tool in BMaps today!
Check out our tutorial on how to get your π-π interactions and hydrogen bonding interactions quantified in real time here or just ask Gibbs!
Let us know which interaction you want to see
automatically quantified next!