DiffDock Diffusion-Based AI Model Available for Quick Protein-Ligand Docking in Boltzmann Maps
Seeking to democratize the latest tooling for computational drug discovery, the Boltzmann Maps team is proud to announce the integration of the AI model DiffDock [1]. In comparison on the PDBBind ligand docking task, DiffDock achieved a 38% top-1 success rate for binding ligands within 2A RMSD of the crystal docking site. This outperformed traditional Glide docking at 23% and other leading deep learning methods at 20% [1].Furthermore, docking runs take less than a minute for most protein-ligand combinations, transforming the possibilities for traditional computational workflows.
Features
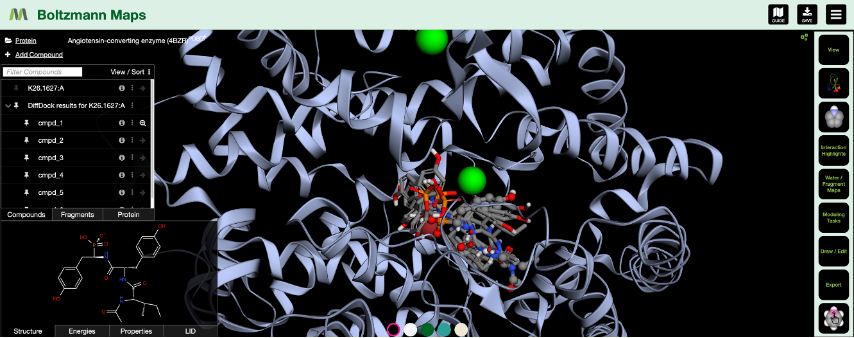
Now available in BMaps as an additional option alongside our previously implemented AutoDock Vina capabilities, DiffDock in Boltzmann Maps now supports:
- Fast rigid, full-protein docking of ligands
- Visualization of up to the 10 best poses for each docked compound
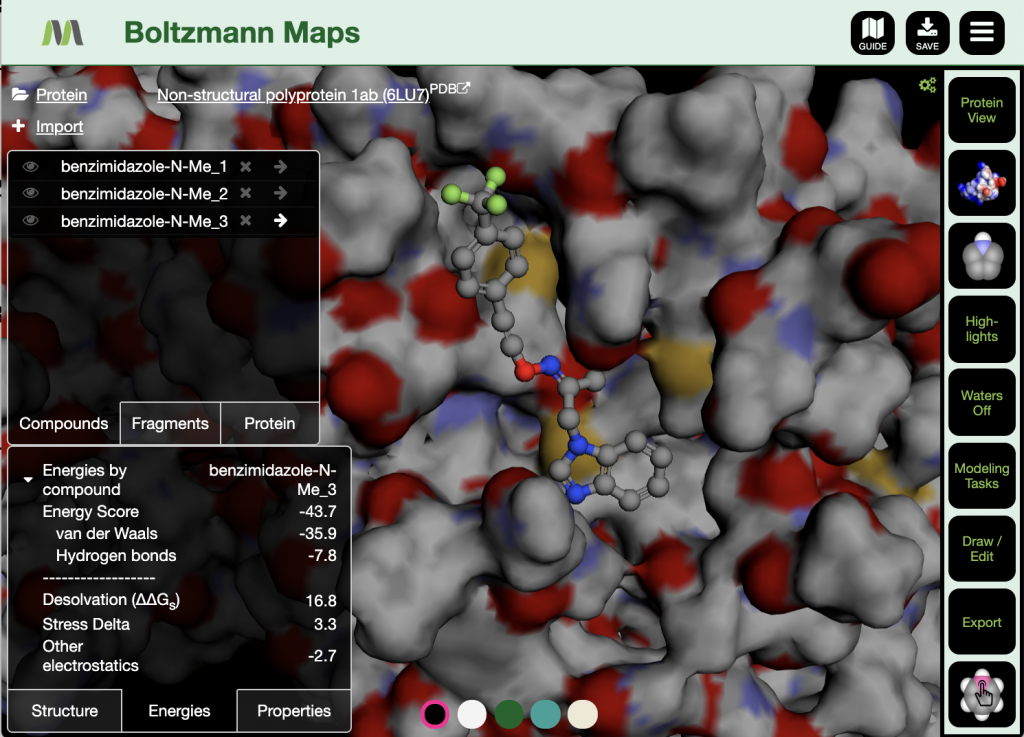
- Energy minimization and scoring of docked poses with calculated physiochemical properties.
Use DiffDock Today!
Freely play around with DiffDock today:
Then, you can follow up your docking runs of multiple compounds by minimizing the docked geometries and calculating the energy score for each pose to discover which compound binds best to your protein of interest! Additionally, you can compare results from AutoDock Vina and hot spot analysis side by side to gain confidence in your results through the use of lateral methods.
Next Steps
This is the first post in a series of blogs on DiffDock.
See the next blog post for a full tutorial on using this tool in BMaps.
For greater detail about the theory and implementation of DiffDock, we recommend the original publication and associated Github. Stay tuned for an upcoming blog post breaking down this theory.
[1] Corso, Gabriele, et al. “Diffdock: Diffusion steps, twists, and turns for molecular docking.” arXiv preprint arXiv:2210.01776(2022).
Need help with your computational chemistry and biology tasks? Conifer Point, maker of Boltzmann Maps, proudly offers CRO services.